
Многоцентровое клиническое исследование — это исследование проводимое по единой программе, но более чем в одном исследовательском центре или клинике.
Статья для тех, кому интересны многоцентровые исследования в рамках регистрации медицинских изделий по правилам ЕАЭС. Мы начнем с разбора официальных Правил, поделимся мнением наших экспертов и партнеров из исследовательских центров. Также будем дополнять статью практическими кейсами и знаниями по мере применения новых правил ЕАЭС.
Содержание
Что такое многоцентровые исследования
Многоцентровые клинические исследования по сути дела обычные клинические исследования, но проводимые не в одном, а в нескольких исследовательских центрах по единому протоколу, и проводимые более чем одним исследователем.
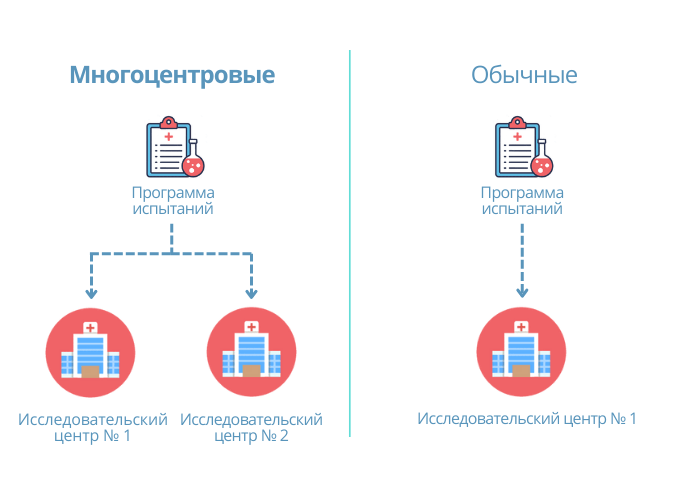
Например, отечественный производитель хочет выпустить на рынок новое изделие — наркозно-дыхательные аппараты. Для этого ему нужно пройти клинические исследования. По новым правилам ЕАЭС для такого изделия потребуется организовать многоцентровые клинические исследования. Это значит, что производитель вместо одного исследовательского центра должен выбрать два. Например, один будет в России, а второй в Беларуси. Ниже расскажем нюансы прохождения таких исследований.
Важный момент, что формат многоцентровых исследований является нововведением по новым правилам ЕАЭС, он становится обязательным для классов риска 3, 2б и имплантируемых медицинских изделий. Дело в том, что раньше по национальным правилам такой формат исследований в рамках регистрации медизделий никогда не применялся, в отличие от лекарственных средств. Поэтому практического опыта применения новых правил для медизделий пока что мало: на начало 2022 в реестре ЕАЭС всего 7 зарегистрированных медизделий. Из них две регистрации проводились в формате многоцентровых клинических исследований.
Кому нужны многоцентровые клинические исследования
В новых правилах ЕАЭС формат клинических исследований напрямую будет зависеть от того какой класс потенциального риска у изделия:
- многоцентровые клинические исследования — для изделий 2б, 3 классов риска и имплантируемых изделий
- обычные клинические исследования — для изделий 1, 2а классов риска
Из-за неправильно установленного класса риска можно составить некорректную программу испытаний, а это повлечет потерю времени и денег. Поэтому важно на первых этапах уделить этому внимание. Подробнее про класс риска можно почитать тут — класс риска [инструмент].
Для тех, у кого изделие не подпадает под многоцентровые исследования, может быть полезно: клинические исследования и особенности регистрации медизделий в ЕАЭС в 2022 году.
Дальше рассмотрим, что делать тем, у кого изделие подпадает под формат многоцентровых исследований.
Этапы многоцентровых исследований
Всего существует два варианта проведения многоцентровых клинических исследований: с участием человека и без. В зависимости от этого будут отличаться этапы:
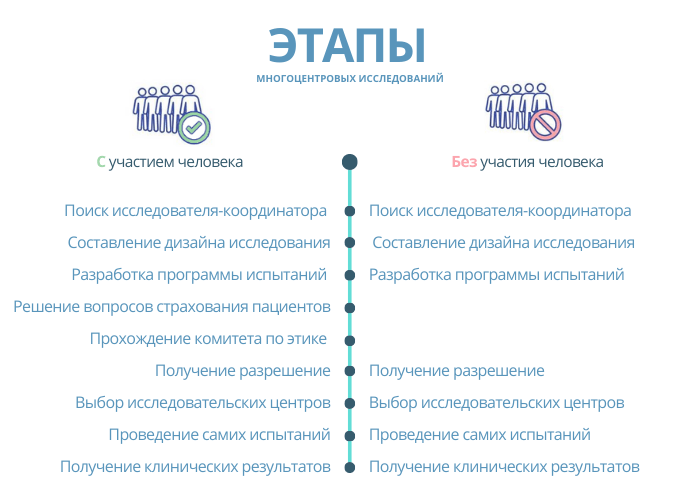
Вариант проведения многоцентровых исследований зависит от класса риска:
- с участием человека: для 3 класса и 2б имплантируемые
- без участия человека: для 2б неимплантируемые
Как подготовиться и на что обратить внимание
Этот раздел будет дополняться по мере применения новых правил ЕАЭС на практике. Обратите внимание, что речь ниже пойдет про медицинские изделия, важно не путать с аналогичными многоцентровыми исследованиями для лекарственных средств.
Расскажем, что уже известно из проведенных многоцентровых исследований. На что обратить внимание производителям на всех этапах:
Найти исследователя-координатора
Для запуска многоцентровых исследований нужно найти исследователя-координатора. Его главная задача координация работы двух исследовательских команд в выбранных клинических центрах. Такого специалиста назначает сам производитель или уполномоченный представитель производителя (УПП). В большинстве случаев такая должность либо уже есть в компании, либо исследователя-координатора могут предоставлять исследовательские центры.
Выбрать исследовательские центры
По регламенту этот этап идет в конце — после получения разрешения на проведение многоцентровых клинических исследований. Но на практике выбрать исследовательские центры можно и в самом начале, важно учитывать нюансы:
- Два исследовательских центра должны быть в разных странах ЕАЭС, например, один в России, а второй в любой стране признания. Обратите внимание на поправки от 18 февраля 2022 года.
- Рекомендуем второй страной для проведения клинических исследований выбирать ту, в которой позже будет согласование заключения экспертизы.
- Один из двух исследовательских центров будет ведущим, он будет вместе с исследователем-координатором агрегировать данные от второго исследовательского центра.
- В отличие от обычных клинических исследований многоцентровой формат затратнее. Два исследовательских центра — каждому оплата за услуги.
Выбрать дизайн исследования
Выше мы уже затронули тему дизайна исследования, например, формат на человеке или нет — это пример дизайна исследования. Если упростить, то на этом этапе определяется проект будущего исследования. По регламенту этим должен заниматься сам производитель, но на практике это совместная работа производителя, исследователя-координатора и исследовательских центров. Именно на этом этапе решается какие методы, инструменты или подходы будут применяться в исследовании.
Разработать программу испытаний
Если дизайн исследования — это проект того, как будут проводиться многоцентровые исследования, то программа испытаний — это документ, который по регламенту составляет сам производитель, но на практике это будет совместная работа производителя, исследователя-координатора и исследовательских центров. Например, один из исследовательских центров разработает программу, потом отправит её на согласование исследователю-координатору, производителю и второму исследовательскому центру. Если нет замечаний, то все согласовывают и подписывают программу. Программа должна быть единой для двух исследовательских команд.
Решить вопрос страхования пациентов
Данный пункт актуален, если многоцентровые исследования проводятся с участием человека. Тогда страхование участников исследования потребуется для прохождения следующих этапов: комитета по этике и получения разрешения РЗН. Нужно будет предоставить копии страховых документов, где указаны условия страхования и компенсации. Важно обратить внимание на то, что страхование осуществляется исходя из законодательств государств, на территории которых проводятся клинические исследования. Условия страхования участников исследований в разных странах ЕАЭС могут отличаться.
Пройти комитет по этике
Следующим важным шагом будет прохождение комитета по этике. Это актуально если исследование с участием человека. Заявитель передает пакет сведений, включающий всё необходимое для полной и тщательной экспертизы планируемого исследования:
- заявление на рассмотрение
- программу планируемого исследования
- индивидуальные регистрационные карты, дневники и вопросники, которые предстоит заполнять исследователям
- описание данных по безопасности медицинского изделия, исследование которого запланировано, а также его технические характеристики, данные проведенных токсикологических испытаний с описанием существующего на данный момент клинического опыта применения медицинского изделия
- брошюру исследователя
- текущую редакцию резюме исследователя и другие материалы, подтверждающие его квалификацию
- материалы, включая рекламные, используемые для привлечения потенциальных субъектов исследования
- форму информированного согласия с описанием процесса его получения и документирования, а также другие формы, содержащие информацию для потенциальных субъектов исследования
- описание всех компенсаций за участие в исследовании для участников исследования, включая покрытие расходов и медицинскую помощь
- информацию об условиях выплат и компенсаций субъектам исследования
- описание условий страхования участников исследования
- положение о согласии следовать этическим принципам, изложенным в соответствующих руководствах
- предыдущие решения, принятые другими комитетами по этике
В каждой стране ЕАЭС действуют свои комитеты по этике. Существуют комитеты на уровне экспертной организации, например есть комитет при Росздравнадзоре, также на уровне самих исследовательских центров. После получения пакета документов комитет:
- рассматривает программу клинических исследований
- оценивает соответствие квалификации исследователя
- дает свое заключение об этической обоснованности/необоснованности
Получить разрешение
На этом этапе уполномоченный орган страны ЕАЭС, где проводятся исследования, в нашем случае Росздравнадзор (РЗН) и уполномоченный орган второй выбранной страны, например в Казахстане принимает решение о возможности проведения исследований. Для этого нужно направить в РЗН следующий пакет документов:
- заявление производителя или его уполномоченного представителя о том, что данное медицинское изделие удовлетворяет применимым требованиям безопасности и эффективности, за исключением свойств и характеристик безопасности и эффективности медицинского изделия, которые должны быть исследованы в ходе клинических исследований, и что по отношению к ним были приняты меры предосторожности для защиты здоровья и безопасности субъектов исследований
- копия заключения комитета по этике, заверенная производителем или его уполномоченным представителем
- брошюра исследователя (согласно приложению N 2)
- образец индивидуальной регистрационной карты субъекта исследования (при наличии)
- технический файл на медизделие, за исключением свойств и характеристик безопасности и эффективности медицинского изделия, которые должны быть определены в ходе клинических исследований (согласно приложению N 3)
- программа клинического исследования с обоснованием количества представляемых медизделий и сроки проведения (согласно приложению N 4)
- перечень неблагоприятных событий, о которых планируется сообщать в РЗН, и указать срок направления такой информации
- копия документа об условиях страхования и компенсаций
Обратите внимание, что если оригиналы документов составлены на иностранном языке, то они предоставляются с переводом на русский язык, заверенный производителем или его уполномоченным представителем.
После получения пакета документов, проверяется комплектность материалов и не позднее 30 рабочих дней направляется решение относительно возможности проведения многоцентровых клинических исследований.
Получить результаты клинических исследований
В конце составляется общий отчет, в который входят результаты обоих исследовательских центров и программа проведенных исследований. Такой отчет официально называют “Акт клинических исследований”, он должен быть подписан исследователями и руководителями медицинских организаций, а также быть утвержден координатором-исследователем.
Нормативные документы:
1. Правила проведения клинических и клинико-лабораторных испытаний (исследований) медицинских изделий
Помощь с многоцентровыми исследованиями
Правила регистрации ЕАЭС сложнее национальных. На начало 2022 в реестре ЕАЭС всего 8 зарегистрированных медизделий, все они — от крупных компаний.
Компания «Новый элемент» уже запустила регистрацию по правилам ЕАЭС стоматологического визиографа и парового стерилизатора. Поэтому, если у вас есть вопросы по регистрации медицинских изделий, обращайтесь за консультацией к нашим экспертам.



